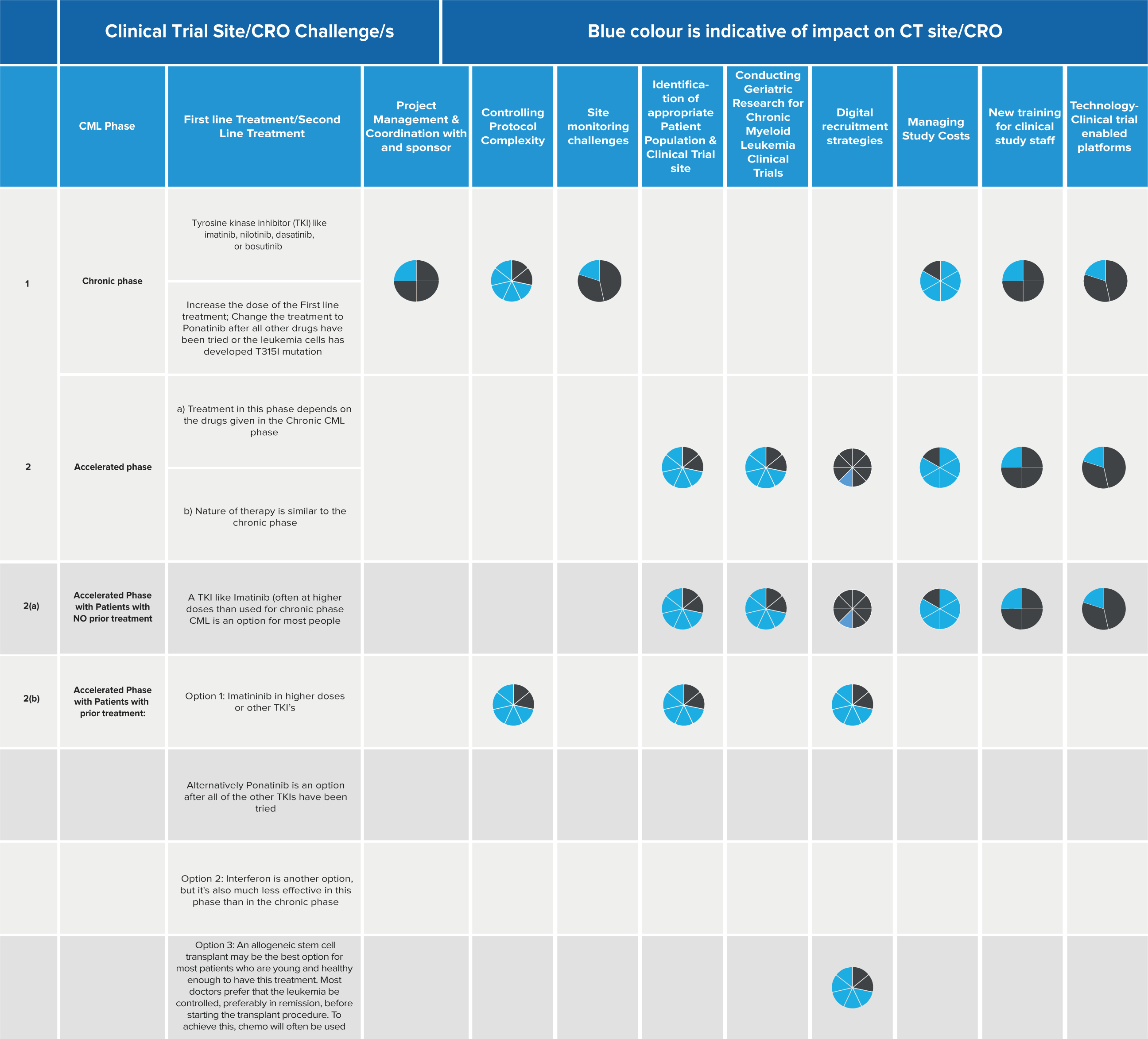
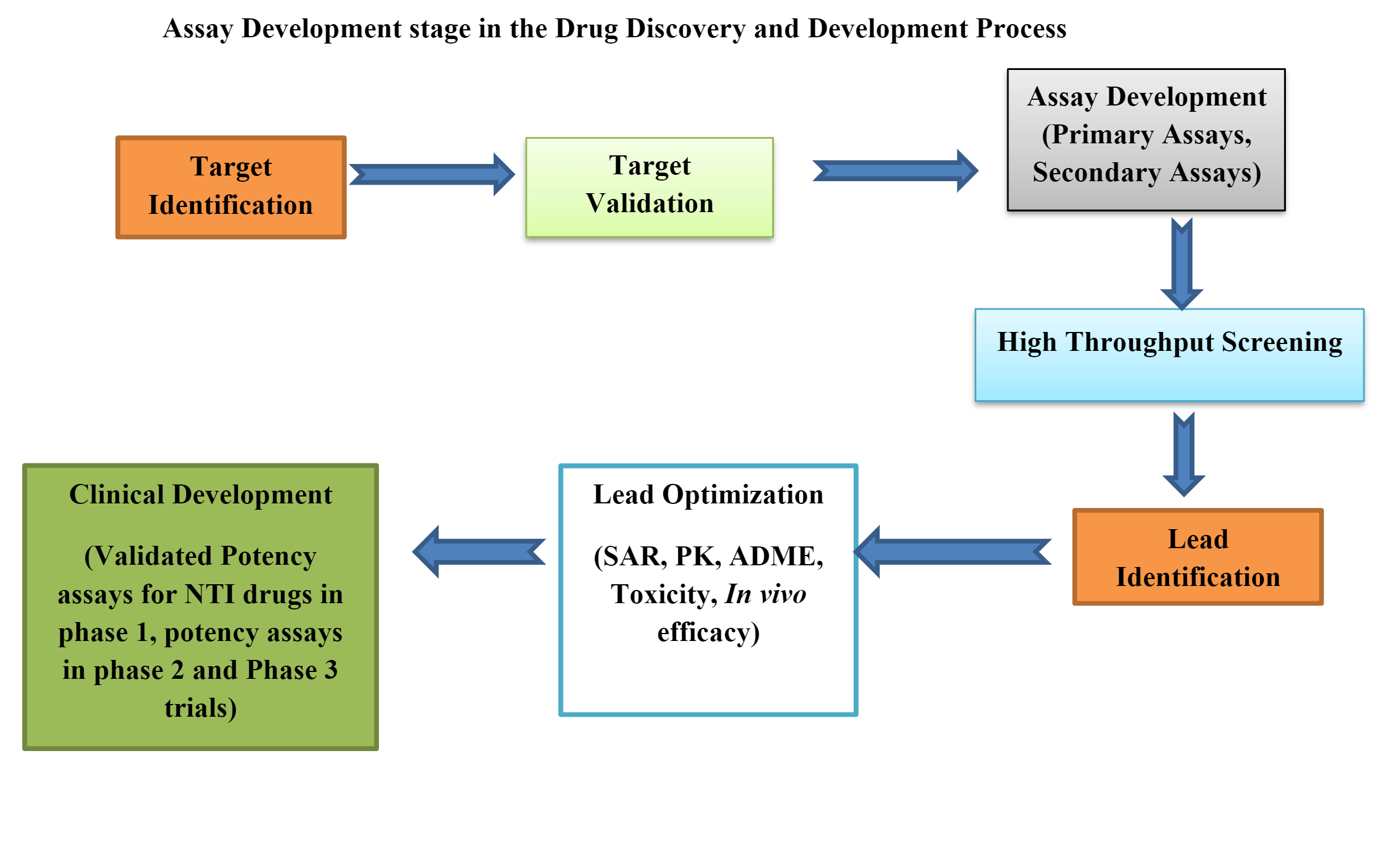
Overview
Pharmacodynamic (PD) biomarkers indicate how a drug affects its target, like a receptor triggering a signalling cascade. They reflect the drug’s impact on the body’s biological or physiological functions. Unlike pharmacokinetics, which focuses on how the body processes a drug, pharmacodynamics explores its effects and mechanisms. These markers are vital in clinical trials, helping assess a drug’s efficacy, safety, and optimal dosage, and in individualizing treatments. They’re crucial in drug development, aiding researchers and healthcare pros in understanding a drug’s interactions and suitability for its intended use. Developing New Chemical Entities (NCE) involves discovering, designing, and synthesizing novel compounds for therapy. Bioanalysis, quantitatively measuring drugs and their metabolites in biological samples, is key in NCE development.
Challenges & Considerations
| Factors | Challenges | Mitigations |
| Analytical Method Development and Validation |
Developing and validating robust bioanalytical methods to quantitate the NCE and its metabolites in complex biological matrices | Rigorously following regulatory guidelines, conducting thorough method validation, and adapting methods as needed during the development process |
| Bio matrix Interference, Matrix Standardization, Sensitivity and Specificity
|
Biological samples like blood or urine might have interfering substances affecting accurate drug measurement. Methods must detect low concentrations and differentiate the drug from other components, while individual differences impact consistency | Efficient sample preparation using surrogate or diverse matrices, optimizing extraction protocols with advanced tools for precision & employing matrix standardization to address inter-individual variability in analysis |
| Automation and Throughput with Emerging Technologies | Maintaining accuracy while meeting high throughput needs. Adopting cutting-edge bioanalytical tech for large molecules, prioritizing contamination control, and addressing ethical considerations with minimal sample volume | Automating processes, streamlining workflows for efficiency and staying updated on new tech; assess their relevance in NCE development with hybrid methods like LBA-MS |
| Integration of Biomarkers |
Incorporating biomarkers into bioanalytical strategies to provide insights into pharmacodynamics |
Exploring and validating biomarkers that align with the pharmacological effects of the NCE |
Strategies for PD Biomarker Quantitation
Quantifying Pharmacodynamic (PD) biomarkers in bioanalysis involves careful planning and execution to ensure accurate and reliable measurement of the biological responses to a drug. Here are the strategies concerning requirements and rationale for PD biomarker quantitation in bioanalysis.
| Requirements | Strategies | Rationale |
| Biomarker Selection and Validation | Choosing PD biomarkers that are relevant, specific, and validated to reflect the pharmacological effects of the drug | Selection based on a strong scientific rationale enhances the likelihood of meaningful results |
| Sample Collection and Processing | Establishing standardized procedures for sample collection and processing to minimize variability | Considering the choice of biological matrices, collection timing, and sample storage conditions |
| Calibration Standards and Quality Control Samples | Preparation of calibration standards with known concentrations of the PD biomarker and including quality control samples | Calibration curves ensure accurate quantitation, while quality control samples assess the precision and accuracy of the assay |
| Internal Standards | Incorporating internal standards into the assay for normalization and to correct for variations | Internal standards help account for analytical variability and matrix effects |
| Validation of Bioanalytical Methods | Rigorously validating bioanalytical methods & following regulatory guidelines | Validate for selectivity, sensitivity, precision, accuracy, linearity, and robustness |
| Use of Stable Isotope-Labeled Internal Standards | Employing stable isotope-labelled internal standards for accurate quantitation | Stable isotope-labelled standards closely mimic the analyte’s behaviour, enhancing precision and accuracy. In the absence of an isotope-labelled internal standard, an analogue IS with similar characteristics can be selected |
| Automation and High-Throughput Techniques | Implementation, automation, and high-throughput techniques for increased efficiency | Automation reduces human error, and high-throughput methods are beneficial in large-scale studies |
| Matrix Effects and Standardization | Addressing matrix effects by standardizing matrices or using matrix-matched standards | Matrix effects can impact accuracy, so careful consideration of matrix standardization is crucial |
Veeda’s Capabilities & Approach for Novel Drug Development Program
Bioanalysis is a vital part of drug development, focusing on accurately measuring drugs and their by-products in biological samples. A successful bioanalysis strategy involves method development, validation, and application in clinical studies.
- At Veeda, method development involves extensive research, considering various factors like drug properties, dose, linearity range, extraction protocols, chromatography, and equipment. Method validation includes experiments ensuring compliance with regulations, such as selectivity, accuracy, precision, sensitivity, matrix effects, and stability studies. In clinical sample analysis, it’s crucial for determining drug levels in biological samples. Incurred sample reanalysis validates reported sample analyte concentrations, ensuring reliability
- Employing emerging technologies like LC-MS/MS machines, ICP-OES, LIMS, and BSL-2 labs enhances our capabilities. Quality management systems (QMS) established protocols ensuring consistent quality standards, customer satisfaction, and regulatory compliance
- Data analysis and statistical approaches at Veeda derive meaningful insights from experimental results, ensuring their reliability and validity
- Regulatory compliance involves adherence to industry-specific laws, guidelines, and standards
- Cross-validation with clinical endpoints ensures alignment between laboratory analyses and clinical outcomes, establishing correlations between measured biomarkers/drug concentrations and therapeutic effects/safety outcomes
Our Expertise in PD Biomarker Method Development & Validation
| Biomarkers | Veeda’s Expertise |
| Alpha-1-acid glycoprotein | Determination of, α1 Acid Glycoprotein (AAG) of in K3EDTA Human Plasma by Using LC-UV with linearity range of 300µg/mL to 5000µg/mL
|
| Coproporphyrin I | Determination of Coproporphyrin I in altered and Unaltered plasma by using LC-ESI-MS/MS, with linearity range of 50pg/mL to 5000pg/mL |
| Symmetric Dimethylarginine (SDMA) | Determination of SDMA in stripped and un-stripped plasma by using LC-ESI-MS/MS, with linearity range of 2.00ng/mL to 4000ng/mL |
| Uridine | Determination of Uridine and L-Dihydroorotic acid(L-DHO) in altered and unaltered, Plasma by using LC-ESI-MS/MS with linearity range of 30ng/ml to 30000ng/ml for Uridine and 3.0ng/mL to 3000ng/mL for LDHO |
| C-peptide | Determination of C-Peptide in human serum by Using ECLIA Method on Immuno-assay Analyzer Cobas e 411 |
Conclusion
Bioanalysis is pivotal in identifying, measuring, and characterizing Pharmacodynamic (PD) markers, which indicate a drug’s biological effects in an organism. Its role involves:
- Identification: Using techniques like mass spectrometry, immunoassays, and chromatography to screen and identify potential PD markers
- Quantification: Developing precise methods to measure PD markers accurately
- PK/PD Modelling: Integrating bioanalytical data into models for predictive insights on drug concentration and PD marker levels
- Dose-Response Assessment: Analyzing concentration-response relationships to establish dose-response curves
- Early Phase Development: Using bioanalytical data to guide decisions about dosing, further development, and safety concerns
- Safety Assessment: Identifying and measuring biomarkers that signal potential safety issues during drug development
Reference:
- Abbas M, Alossaimi MA, Altamimi AS, Alajaji M, Watson DG, Shah SI, Shah Y, Anwar MS. Determination of α1-acid glycoprotein (AGP) concentration by HPLC in patients following local infiltration analgesia for primary total hip arthroplasty and its relation to ropivacaine (total and unbound). Frontiers in Pharmacology. 2023;14
- Kandoussi H, Zeng J, Shah K, Paterson P, Santockyte R, Kadiyala P, Shen H, Shipkova P, Langish R, Burrrell R, Easter J. UHPLC–MS/MS bioanalysis of human plasma coproporphyrins as potential biomarkers for organic anion-transporting polypeptide-mediated drug interactions. Bioanalysis. 2018 May;10(9):633-44
- Shin S, Fung SM, Mohan S, Fung HL. Simultaneous bioanalysis of l-arginine, l-citrulline, and dimethylarginines by LC–MS/MS. Journal of Chromatography B. 2011 Mar 1;879(7-8):467-74
- Yin F, Ling Y, Martin J, Narayanaswamy R, McIntosh L, Li F, Liu G. Quantitation of uridine and L-dihydroorotic acid in human plasma by LC–MS/MS using a surrogate matrix approach. Journal of Pharmaceutical and Biomedical Analysis. 2021 Jan 5;192:113669
- US Food and Drug Administration; U.S. Department of Health and HumanServices; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM). Bioanalytical Method Validation: Guidance for Industry; U.S. Department of Health and Human Services, Food and Drug Administration: Silver Spring, MD, 2018